
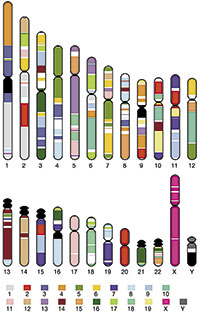
Figura 1. Comparación entre los cromosomas del ratón y del hombre. Mediante el código de colores, que corresponden a los cromosomas del ratón indicados en la parte inferior, puede apreciarse a qué fragmentos de los cromosomas humanos son equivalentes, entendiendo como tal la conservación de los genes contenidos en los mismos. Queda de manifiesto cómo desde la separación de los linajes de roedores y humanos se han producido numerosos cambios en la disposición cromosómica, pero el contenido total en genes de ambos organismos es muy semejante.
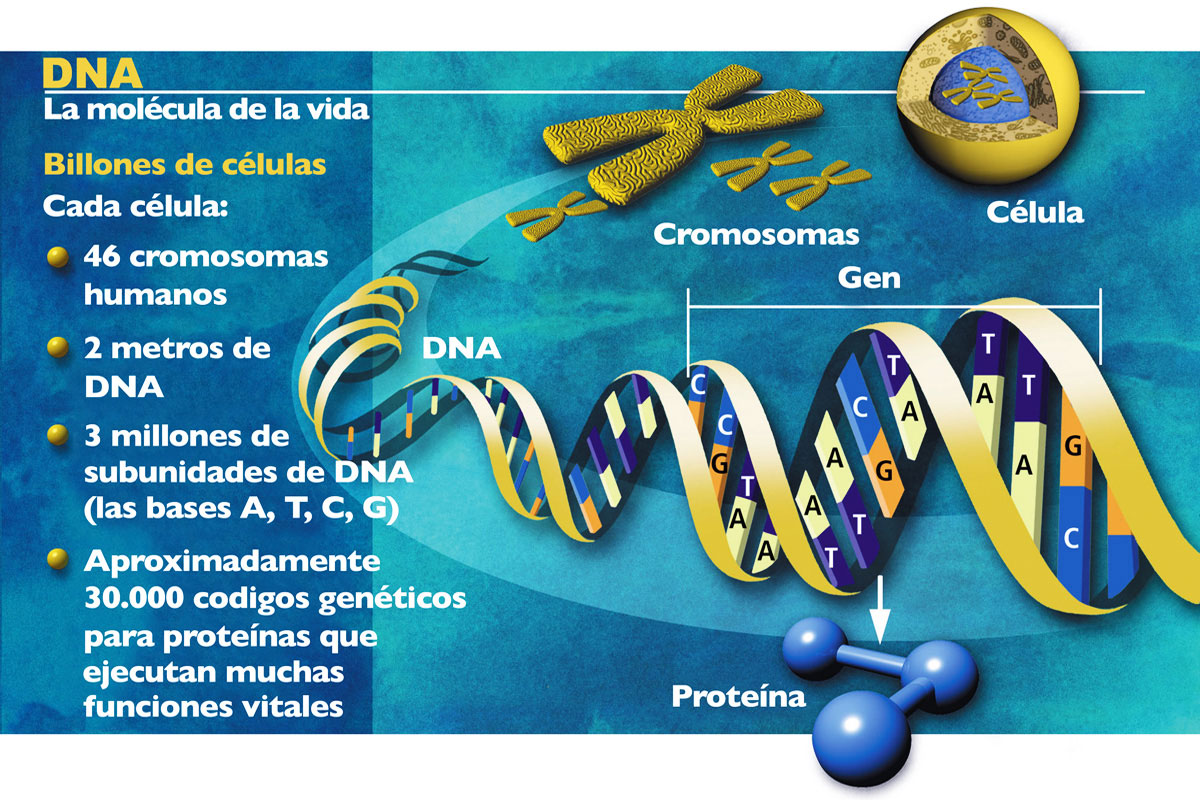
El genoma es el conjunto del material hereditario de un organismo, la secuencia de nucleótidos que especifican las instrucciones genéticas para el desarrollo y funcionamiento del mismo y que son transmitidas de generación en generación, de padres a hijos. En él, además de los genes propiamente dichos, se incluyen regiones espaciadoras, regiones reguladoras, restos de genes antaño funcionales y muchas otras secuencias de función o papel todavía desconocido, si es que tienen alguno. De hecho, en el genoma humano, apenas el 1,5% del material hereditario tiene una función codificante, es decir, corresponde a lo que solemos entender por genes. Por tanto, el genoma de un organismo es el depositario de la información que permite que cada organismo se desarrolle y responda a las exigencias impuestas por el medio. Pero, además, el genoma es depositario de los cambios que, a lo largo de la historia de la especie correspondiente y de todas sus antecesoras, han permitido su supervivencia hasta nuestros días. En consecuencia, en el genoma se almacena información de dos tipos: una de inmediata utilidad para el organismo y otra que sirve como registro histórico de éste y de sus ancestros (fig. 1). Ambos tipos de información son explotados por la biología actual, tanto en su vertiente funcional como en la histórica o evolutiva.
Alrededor del genoma se plantean varias cuestiones que conviene aclarar. La primera tiene que ver con su capacidad para determinar total o parcialmente el funcionamiento del organismo. A modo de analogía, podríamos comparar el genoma con los planos de una casa elaborados por un arquitecto en su estudio. El resultado final depende de muchas decisiones e intermediaciones no siempre previsibles: la disponibilidad de materiales en cada momento, la interpretación realizada por el director de obra, la solución adoptada ante algún imprevisto, las modificaciones introducidas por el propietario, ¡hasta las preferencias estéticas de éstos! Por tanto, el edificio final puede diferir del imaginado inicialmente por el arquitecto que lo planeó, pero estas diferencias se producen más fácilmente en detalles accesorios y menos en los fundamentales. Igualmente, podemos decir que el genoma de un organismo contiene un conjunto de instrucciones, pero que la forma en que éstas son llevadas a cabo depende a su vez de contingencias ambientales e históricas que pueden llevar a diferencias entre planos (o fragmentos de ellos) en principio iguales. En consecuencia, la naturaleza de las instrucciones genéticas no es completamente determinista en todos los casos, si bien hay una serie de procesos en los que sí se cumple esa perfecta relación entre herencia y expresión final.
«Del genoma humano, a penas el 1,5% del material hereditario tiene una función codificante, es decir, corresponde a lo que solemos entender por genes»
Otra cuestión que debemos aclarar es que no existe una relación uno a uno entre genes y caracteres observables o, al menos, que esta relación dista mucho de ser general. En algunas ocasiones, un único gen determina un carácter completamente: por ejemplo, el sistema sanguíneo ABO o el grupo Rh son determinados por un solo gen, respectivamente. Esta misma situación se presenta con ciertas alteraciones genéticas y el desarrollo de patologías, lo que facilita enormemente el diagnóstico precoz y abre las posibilidades para la terapia genética. Pero muchos caracteres, la gran mayoría, incluyendo muchas condiciones de interés para la medicina o la psicología, tienen una base poligénica, es decir, no existe “el gen” que determina el carácter de forma unívoca, sino que éste es el resultado de la acción simultánea de muchos genes, en ocasiones centenares de ellos, no todos con la misma participación y sobre los cuales hay que añadir el efecto del ambiente antes comentado. Muchos de los debates clásicos sobre el determinismo genético de los rasgos de la conducta y la personalidad, o la inteligencia, surgen de una incorrecta apreciación de esta naturaleza dual de la expresión de los caracteres.
Finalmente, y como sistemas complejos que somos todos los seres vivos, debemos considerar el papel que tienen las interacciones entre las fracciones componentes del genoma a la hora de especificar el resultado final, al menos en su componente genético. Sabemos que un gen puede afectar a más de un carácter y que un carácter puede ser afectado por más de un gen. Por tanto, una modificación en un gen puede provocar alteraciones en varios caracteres, lo que conocemos como efectos pleiotrópicos, y la expresión de cierto carácter puede depender de qué variantes se encuentran presentes en dos o más genes diferentes, dando lugar al fenómeno conocido como epistasia. Dado el número de genes presentes en cualquier organismo (del orden de decenas de miles para los animales y plantas), se abre un tremendo abanico de posibilidades de interacción entre dos o más genes. Actualmente carecemos de siquiera una idea aproximada del papel que desempeñan las epistasias en la gran mayoría de caracteres fenotípicos.
«La gran mayoría de caracteres tiene una base poligénica, es decir, no existe el «gen» que determina el carácter de forma unívoca, sino que éste es el resultado de la acción simultánea de muchos genes»
¿Cómo se estudian los genomas?
Si bien la secuenciación del genoma humano marca un hito en la biología, las contribuciones desde otras disciplinas científicas han sido imprescindibles para lograrlo. El proyecto se ha beneficiado de avances en la química, la física, las matemáticas, la informática, y ha dado lugar, incluso, al nacimiento de una nueva disciplina integradora, la bioinformática, sin la cual no se hubiese podido culminar, como veremos posteriormente. No nos puede extrañar, en consecuencia, que entre los integrantes de los equipos que han logrado completar la secuencia del genoma humano se encuentren científicos de disciplinas muy dispares. Esto mismo se repite en los equipos que han logrado descifrar las secuencias de otros organismos. El estudio del genoma en su integridad es el resultado de avances técnicos y conceptuales que empezaron hace unos 25 años, cuando Fred Sanger y Walter Gilbert desarrollaron sendos métodos para obtener la secuencia de nucleótidos de un fragmento pequeño de DNA. Al poco, el primero de ellos consiguió secuenciar el genoma de un bacteriófago, un virus que infecta células bacterianas, el fX174. Inmediatamente empezaron a acumularse secuencias de genomas víricos, seguidas, a principios de los años 80, por la secuencia del genoma mitocondrial humano. La obtención de cada una de estas secuencias representaba la culminación de una ardua tarea de varios meses, incluso años, a pesar de que estos genomas sólo contienen unos pocos miles de nucleótidos. Dado que el genoma humano consta de unos 3.000 millones de nucleótidos, el salto que representa pasar de secuenciar un genoma viral al humano parecía infranqueable en 1980, cuando David Botstein y colaboradores propusieron este objetivo a la comunidad científica. El intervalo de casi una década entre la propuesta inicial y la decisión política final de llevarla a cabo permitió que se produjesen avances decisivos en varios frentes, como en las técnicas de mapeo físico, que permiten el aislamiento de genes y regiones concretas a partir de su localización cromosómica, y en las estrategias de secuenciación mediante “perdigonadas” aleatorias, que posteriormente permitieron automatizar un proceso hasta entonces manual.

Figura 2. Gráfico temporal mostrando los avances en la secuenciación de genomas completos. En la actualidad el número de proyectos de secuenciación de genomas completos en marcha supera los dos centenares, a los que cabría añadir otros muchos que se llevan a cabo en empresas privadas y que no son hechos públicos a la comunidad.
A finales de 1990 se lanzó el Proyecto Genoma Humano con la creación de centros de secuenciación en Estados Unidos, Reino Unido, Francia y Japón y con el apoyo de la Comunidad Europea. En los cinco años siguientes se progresó rápidamente en dos frentes: la construcción de mapas genéticos y físicos de los genomas humano y de ratón, lo que facilitó herramientas indispensables para la identificación de genes ligados a enfermedades y la fijación de hitos para la secuenciación posterior, y en la secuenciación de dos genomas de organismos eucariotas, la levadura Saccharomyces cerevisiae, empleada para obtener el pan y la cerveza, entre otros alimentos, y el gusano nematodo Caenorhabditis elegans (fig. 2). En ese momento, la estrategia favorita para la secuenciación del genoma humano se desarrollaba en dos fases (fig. 3). En la primera (fig. 4), el genoma se dividía en fragmentos de tamaño adecuado (de sólo unos centenares de miles de bases) que eran, a su vez, secuenciados mediante la estrategia de la perdigonada. Para fragmentos de este tamaño, la estrategia de la perdigonada necesita que cada nucleótido sea secuenciado varias veces, cuantificadas con el factor de cobertura o redundancia, con el objetivo de que no queden regiones del fragmento sin secuenciar al menos una vez. A medida que el fragmento es mayor en tamaño, hay una tendencia a no aumentar la redundancia tanto como es necesario para garantizar que ningún nucleótido queda sin secuenciar, por lo que habrá algunos huecos en la secuencia final del mismo. De igual manera, para la obtención de esos fragmentos de algunos centenares de miles de bases, se seguía una estrategia parecida, por lo que cada región genómica debía estar representada varias veces si no se quería dejar alguna de ellas sin analizar. La segunda fase completaba la estrategia ciega de la perdigonada buscando llenar aquellos posibles huecos faltantes en los dos niveles mencionados. Esta estrategia fue la adoptada por el consorcio público y debía producir una primera secuencia hacia 2003.
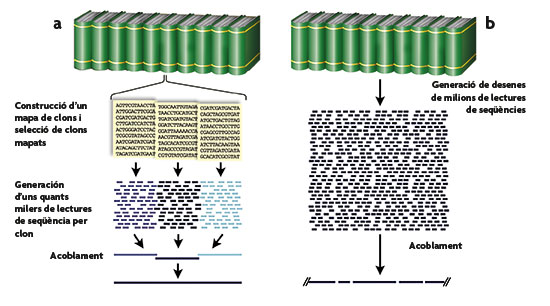
Figura 3. Comparación entre las dos estrategias de secuenciación del genoma humano, la seguida por el consorcio público (a) y la del grupo liderado por Craig Venter (b). En la primera de ellas, detallada con más precisión en la figura 4, se parte de una colección ordenada, mediante mapeo físico y genético, de fragmentos de tamaño mediano, que son secuenciados por el procedimiento de la perdigonada. Por el contrario, en la segunda estrategia se procede a la secuenciación completa del genoma por el procedimiento de perdigonada, con un nivel elevado de redundancia para garantizar la lectura de todas las posiciones, quedando para poderosos sistemas informáticos la tarea de ensamblar las secuencias fragmentarias obtenidas en una única secuencia lineal. En ambas estrategias se plantean problemas con las zonas del genoma ricas en DNA repetitivo, si bien en la primera es más fácil identificar y cuantificar la extensión de las zonas no secuenciadas por este motivo.
En 1998, Craig Venter, previamente uno de los líderes del consorcio público y entonces ya en el sector privado, lanzó el desafío que condujo finalmente a la aceleración de todo el proceso. Su propuesta consistía en abandonar las fases de mapeo físico y ordenación de los clones con fragmentos grandes para pasar directamente a la secuenciación completa del genoma mediante el método de perdigonada, dejando que nuevos algoritmos y ordenadores más potentes se encargasen del ensamblaje de toda la secuencia. Recibida con escepticismo, al menos, su estrategia demostró su capacidad de secuenciar un genoma complejo en un tiempo récord al lograr secuenciar el genoma (180 millones de nucleótidos) de Drosophila melanogaster, la mosca del vinagre y organismo modelo de los genéticos desde principios del siglo XX, en apenas un año. Su grupo comenzó la secuenciación del genoma humano (de hecho, el genoma de 5 individuos diferentes) el 8 de septiembre de 1999 y concluyó la fase de obtención de datos el 17 de junio de 2000. El ensamblaje sobre el que se basó la publicación, simultánea con el resultado del consorcio público el 15 de febrero de 2001, se completó en 3 meses y medio.
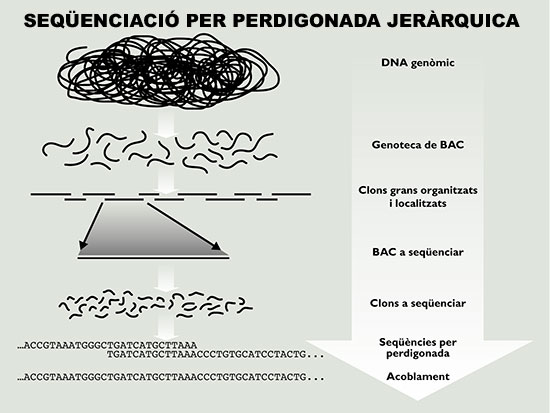
Figura 4. Esquema de la estrategia de secuenciación por perdigonada jerárquica, la empleada por el consorcio público. Se construye una genoteca fragmentando el genoma (o cromosoma) deseado y clonándolo en vectores que aceptan insertos grandes (en este caso cromosomas bacterianos artificiales o BAC). Los fragmentos de DNA genómico presentes en la genoteca se organizan en un mapa físico y a continuación se eligen los clones BAC individuales para su secuenciación por perdigonada. Por último, las secuencias de los clones se ensamblan para reconstruir la secuencia del genoma.
A lo largo del casi año completo que ha transcurrido desde la publicación, han aparecido numerosos estudios basados en el análisis comparativo de ambas secuencias del genoma humano, a la vez que, especialmente algunos investigadores pertenecientes al consorcio público, han suscitado serias dudas sobre la factibilidad de la secuencia de la perdigonada para un genoma tan grande como el humano si no hubiese tenido acceso, el grupo liderado por C. Venter, a la información publicada por el consorcio público. Entre los estudios comparativos, sorprende que, a pesar de la buena coincidencia respecto del número de genes previstos por ambos grupos (en torno a los 35.000), buena parte de ellos no coinciden entre las dos predicciones. Por otra parte, algunas afirmaciones, como la existencia de unos 150 genes en el genoma humano más próximos a genes de procariotas, bacterias en este caso, que a ningún otro organismo evolutivamente más emparentado con nosotros, han sido claramente refutadas.
A pesar de lo mucho que hemos aprendido, y la rapidez con que lo hemos logrado, aún quedan muchas incógnitas por resolver, muchas sorpresas con las que replantearnos nuestra concepción actual y muchos detalles por revelar antes de poder utilizar provechosamente la inmensa cantidad de información que encierra el genoma. Sólo entonces estaremos en condiciones de comprender, a todos los niveles, cómo funciona un ser vivo.
Las revistas Nature y Science han depositado gran cantidad de inforamción libremente accesible en sus servidores de Internet:




