Las proteasas del coronavirus: un objetivo prioritario en la búsqueda de antivirales

Si algo ha quedado claro en la actual pandemia protagonizada por la enfermedad COVID-19, es que todo ha ocurrido a una velocidad vertiginosa, también en la ciencia. Desde que el pasado 31 de diciembre de 2019 las autoridades de la República China alertaron a la Organización Mundial de la Salud de que estaban tratando decenas de casos de neumonía de origen desconocido, científicos de todo el planeta se pusieron manos a la obra para primero, descubrir el patógeno que se ocultaba detrás de estas neumonías, y después, estudiarlo con detalle para poder desarrollar alguna estrategia terapéutica que mitigara sus devastadores efectos.
Así, en las primeras semanas de enero un grupo de investigadores de la Universidad de Fudan (China) secuenció el genoma del primer SARS-CoV-2, el coronavirus responsable de la nueva enfermedad, a partir de muestras del fluido de lavado broncoalveolar de un paciente de 41 años sin historial clínico de hepatitis, tuberculosis o diabetes, y lo liberó incluso antes de que fuese aceptado para su publicación el 28 de enero (Nature 580, E7 [2020].). A partir de ese momento, investigadores de todo el mundo iniciaron una carrera trepidante para conocer los detalles moleculares de este patógeno. En la actualidad, solo unos pocos meses después y con varios miles de genomas secuenciados, se ha generado una cantidad ingente de información ciertamente difícil de manejar.
Organización y expresión del genoma
El genoma del SARS-CoV-2 está formado por un RNA de cadena sencilla de alrededor de 29.900 nucleótidos que puede ser reconocido y traducido por la maquinaria celular de síntesis de proteínas de las células infectadas. Este genoma puede codificar hasta 29 proteínas, si bien no está claro todavía si todas estas proteínas se sintetizan realmente durante el ciclo vital del virus. De las proteínas codificadas en el genoma cuatro de ellas dan forma a la estructura del coronavirus y por ello se denominan proteínas estructurales. Estas cuatro proteínas estructurales son la glicoproteína de la superficie (Spike, S) que forma las espículas características de esta familia de virus; la proteína que forma la matriz de la membrana (Membrane, M) que es la más abundante en la envuelta lipídica del virus; una pequeña proteína también embebida en la membrana lipídica que se encuentra en un número reducido de copias (Envelope, E), y la proteína que se asocia al RNA viral estabilizándolo y que forma la nucleocápside del virus (Nucleocapsid, N).
«Si alguna cosa ha quedado clara en la actual pandemia protagonizada por la enfermedad COVID-19, es que todo ha ocurrido a una velocidad vertiginosa, también en la ciencia»
Estas cuatro proteínas estructurales están codificadas en el extremo 3’ del genoma (Figura 1) y, al igual que otras nueve proteínas accesorias codificadas en esta región, no son traducidas directamente por los ribosomas de la célula infectada, sino que se sintetizan a partir de RNA subgenómicos (sgRNA) generados por una proteína viral encargada de replicar (copiar) el genoma del virus (nsp12, ver abajo). En la actualidad todavía no conocemos si todas estas proteínas accesorias se sintetizan en los pacientes CoVid19, y de hecho los estudios más recientes no han conseguido detectar todos los sgRNA (Kim et al., The Architecture of SARS-CoV-2 Transcriptome, Cell [2020].) y tampoco hay evidencias de la presencia de algunas de estas proteínas en las células infectadas.
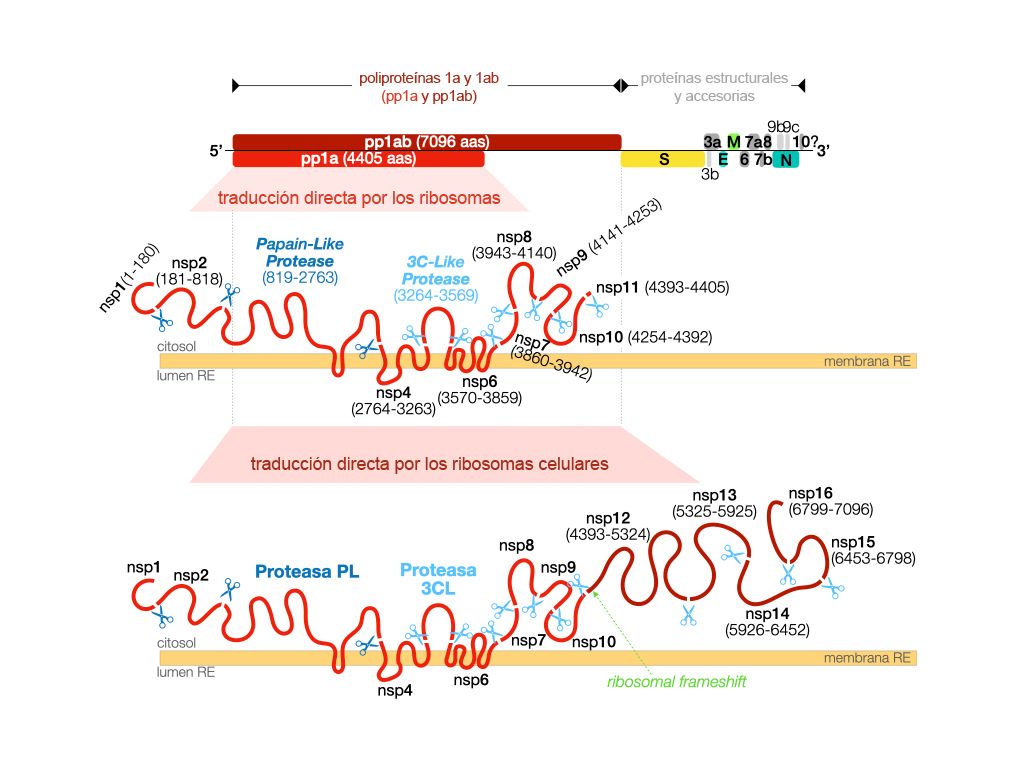
Figura 1. Representación esquemática de la organización del genoma del SARS-CoV-2 y de las proteínas que codifica, ilustrándose con mayor detalle las poliproteínas pp1a y pp1ab que contienen las proteasas PL y 3CL.
A diferencia de lo que ocurre con las proteínas estructurales y accesorias, que necesitan los sgRNAs como ‘mensajeros’ para su síntesis, el resto del genoma del coronavirus codifica en su extremo 5’ (lo que no es casual) dos poliproteínas que sí pueden ser sintetizadas directamente a partir del RNA genómico por los ribosomas de la célula infectada. Esto quiere decir que esta región, que representa aproximadamente el 67% del genoma del virus, se organiza como dos pautas de lectura abiertas (ORF1a y ORF1ab, véase la Figura 1) que codifican varias proteínas que no están separadas entre ellas, sino que se sintetizan como largas cadenas de aminoácidos que deberán ser digeridas (cortadas) para generar 16 proteínas no estructurales (non-structural proteins, nsp). Como veremos, este hecho diferencial en el ciclo del virus constituye uno de sus posibles talones de Aquiles.
Las regiones codificantes de las ORF1a y ORF1ab comparten la misma información en su extremo 5’, de forma que cuando los ribosomas se unen al RNA viral sintetizan la poliproteína de membrana 1a (polyprotein1a, pp1a) en el entorno del retículo endoplásmico de la célula infectada. Como hemos señalado, esta gran poliproteína de 4405 aminoácidos debe cortarse para dar lugar a las proteínas funcionales del virus. Para ello deben romperse los enlaces químicos, enlaces peptídicos, que conforman el esqueleto de las proteínas. En condiciones normales este proceso es extraordinariamente lento, lo que explica que las proteínas sean estables en los seres vivos (el tiempo necesario para la rotura de enlaces peptídicos a temperatura ambiente es de varios cientos de años).
«Para poder desarrollar fármacos antivirales eficaces es necesario conocer con detalle la estructura y el mecanismo de acción de las proteasas»
Para llevar a cabo esta rotura a una velocidad compatible con el ciclo de replicación del virus, es necesaria la acción de enzimas, unas proteínas especializadas en acelerar reacciones químicas. El virus codifica dos de estas proteínas que reciben su nombre por la función que realizan: proteasas, rotura de proteínas. La principal de estas proteasas, la 3CL (3Chymotrypsin-Like), forma parte de la pp1a y una vez sintetizada se autoproteoliza (se corta a sí misma) y en su forma madura realiza 5 cortes adicionales en la secuencia de la poliproteína para generar las proteínas no estructurales nsp4-nsp11. Además de esta proteasa, los coronavirus codifican otra denominada PL (Papain-Like), que dificulta la respuesta celular frente al virus y realiza 3 cortes en la pp1a generando las nsp1, nsp2 y a sí misma.
El final de la secuencia que codifica la pp1a presenta una señal (un codón) de parada que indica al ribosoma el fin de la síntesis de la poliproteína. Sin embargo, en aproximadamente el 60% de los casos, los ribosomas al traducir esta región del RNA viral pueden sufrir un cambio en la pauta de lectura (ribosomal frameshift), desapareciendo de esta forma la señal de parada. Este cambio permite a los ribosomas leer más allá de la región que codifica la pp1a, lo que conduce a la síntesis de la pp1ab. La pp1ab es una poliproteína enorme (7096 aminoácidos) que incluye además de las proteínas no estructurales descritas en la pp1a entre otras a la enzima que copia el genoma del virus (RNA polimerasa RNA dependiente, nsp12), de la que hemos hablado anteriormente.
Inhibiendo las proteasas
Como podemos imaginar, la actividad de ambas proteasas resulta crucial para la replicación del virus, por lo que han emergido como unas de las dianas preferentes para el desarrollo de fármacos que nos permitan tratar esta terrible enfermedad. Para poder desarrollar fármacos antivirales eficaces es necesario conocer con detalle la estructura y el mecanismo de acción de las proteasas. De nuevo en tiempo récord (¡en enero!) dispusimos de las primeras estructuras de alta resolución de la proteasa 3CL (Science 24 Apr 2020:Vol. 368, Issue 6489, pp. 409-412; y Nature [2020].). La rapidez de este notable éxito por parte de dos grupos de investigación de forma independiente (uno en Berlín y otro en Shanghai) se debió en gran parte a la experiencia que ambos grupos adquirieron trabajando con el coronavirus SARS-CoV, causante de la epidemia del síndrome respiratorio agudo grave que apareció en el sudeste asiático entre los años 2002 y 2004. Las proteínas del SARS-CoV-2 presentan una gran similitud con las de este otro coronavirus (hasta el 96% en el caso de la proteasa principal) y con las del coronavirus causante del brote del síndrome respiratorio de Oriente Medio (MERS).
Resulta cuando menos curioso que líneas de investigación que podrían haberse considerado de poca actualidad hace solo unos meses debido a la desaparición de los dos brotes mencionados, hayan resultado ahora claves para poder abordar de una forma rápida el estudio del SARS-CoV-2. Este hecho nos advierte, una vez más, del peligro de que la ciencia se oriente únicamente por criterios de aparente utilidad o aplicabilidad inmediata.
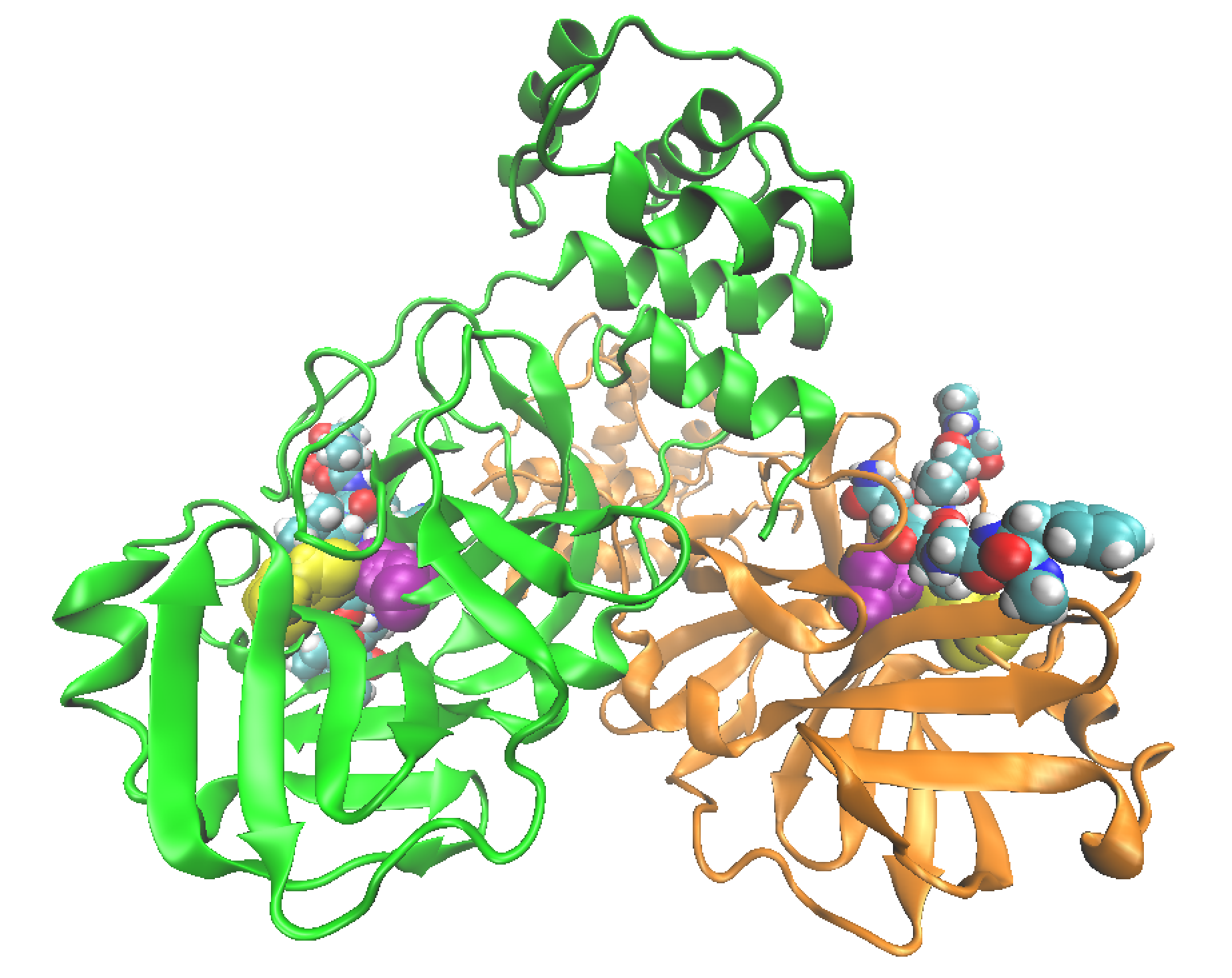
Figura 2. Representación de la estructura de la proteasa 3CL-Like. Cada uno de los protómeros aparece representado en un color con un péptido sustrato (representado en forma de esferas para cada uno de sus átomos) en cada uno de los centros activos. La cisteína e histidina catalíticas aparecen representadas como esferas en color amarillo y púrpura respectivamente en cada uno de los protómeros.
La 3C-Like proteasa del virus es un dímero, formado por dos subunidades idénticas, tal y como puede observarse en la Figura 2. La formación del dímero es esencial para su actividad, ya que el centro activo, el lugar concreto donde la proteasa se une con la poliproteína sustrato y lleva a cabo su actividad, queda conformado completamente cuando se produce la interacción entre las dos subunidades. Cada uno de los dos centros activos del dímero contiene una pareja de aminoácidos encargados de llevar a cabo la rotura de enlaces peptídicos en las poliproteínas, la diada catalítica formada por una cisteína, Cys145, y una histidina, His41 (véase el vídeo adjunto).
La proteasa principal del virus presenta una gran especificidad para romper el enlace peptídico formado entre un residuo de glutamina y otro aminoácido de tamaño pequeño (glicina, alanina o serina). Ninguna de las proteasas conocidas en humanos presenta esta especificidad, lo que convierte a la proteasa principal del SARS-CoV-2 en una diana terapéutica excelente, ya que un fármaco diseñado para inhibir su actividad tendrá bajas probabilidades de interaccionar con proteasas humanas y por tanto de presentar efectos secundarios. Estos inhibidores son moléculas capaces de unirse al centro activo de la proteasa impidiendo el acceso de las poliproteínas y que, por tanto, la proteasa realice su función, lo que implica la interrupción del ciclo de replicación del virus. De hecho, la inhibición de proteasas principales de virus es ya una estrategia eficiente que se usa en el tratamiento de otras epidemias, como es el caso del SIDA.
Como hemos señalado antes, el diseño de inhibidores para la 3C-Like proteasa del SARS-CoV-2 se está llevando a cabo partiendo del conocimiento adquirido sobre la inhibición de su enzima equivalente en el SARS-CoV. El grupo de Berlín (Science 24 Apr 2020:Vol. 368, Issue 6489, pp. 409-412) utilizó la información estructural obtenida para la proteasa principal del SARS-CoV-2 para optimizar un inhibidor que ya se había mostrado efectivo contra las proteasas principales del SARS-CoV y del MERS-CoV. Este inhibidor es capaz de unirse de forma irreversible al centro activo de la proteasa, formando un enlace estable con la cisteína catalítica, dejándola de esta forma inservible para realizar su función sobre las poliproteínas.
Por su parte, el grupo de Shanghai obtuvo la estructura de la proteasa principal junto con un inhibidor distinto unido también a la cisteína catalítica (Nature. [2020].). Este inhibidor se había desarrollado también contra las proteasas principales del SARS-CoV y del MERS-CoV. Además, el equipo de Shanghai realizó un rastreo entre fármacos conocidos y productos naturales con el objetivo de seleccionar otros posibles puntos de partida para desarrollar inhibidores más eficaces. Así, seleccionaron 30 candidatos cuyas estructuras en complejo con la proteasa se están resolviendo en el Diamond Light Source, un laboratorio sincrotón situado en el Reino Unido. Este laboratorio ha puesto en marcha una interesante iniciativa haciendo accesibles al público todas las estructuras de complejos proteasa-inhibidor resueltas y abriendo una página web en la que es posible sugerir posibles inhibidores que, tras ser evaluados mediante inteligencia artificial, son ensayados tras priorizarse en base a criterios tales como su facilidad de síntesis o su posible toxicidad.
«La obtención de un fármaco capaz de inhibir alguna de las proteasas del SARS-CoV-2, o de cualquier otro agente patógeno, es un proceso largo y complejo que requiere de la participación de diferentes ramas de la ciencia»
La proteasa PL (Papain-Like) contiene también en su centro activo una cisteína y una histidina encargadas de llevar a cabo de rotura del enlace peptídico. Por tanto, el mecanismo empleado para actuar como proteasa es muy similar al empleado por la 3C-Like. Sin embargo, la proteasa PL actúa específicamente sobre enlaces peptídicos situados tras dos glicinas consecutivas, usando una secuencia de reconocimiento que, desafortunadamente, también es empleada por algunas proteasas humanas. Esta coincidencia de secuencia de reconocimiento hace más difícil el diseño de inhibidores específicos para la proteasa PL del virus que no causen, a su vez, efectos secundarios al interferir también la actividad de proteasas humanas. De nuevo en este caso los estudios realizados sobre la proteasa PL del SARS-CoV o del MERS-CoV, muy similares a la del SARS-CoV-2, pueden facilitar el desarrollo de inhibidores específicos para esta proteasa.
A pesar de todos los logros alcanzados en tan corto plazo de tiempo, es necesario subrayar que la obtención de un fármaco capaz de inhibir alguna de las proteasas del SARS-CoV-2, o de cualquier otro agente patógeno, es un proceso largo y complejo que requiere de la participación de diferentes ramas de la ciencia. El proceso puede comenzar por la determinación de la estructura de la proteína diana, seguido del diseño asistido por ordenador de moléculas candidatas a inhibir su actividad, la síntesis química, la realización de pruebas bioquímicas sobre su actividad in vitro y, finalmente, el estudio de la viabilidad como fármaco administrado primero a células, luego a animales de experimentación y finalmente a humanos. Este último paso es fundamental para comprobar los posibles efectos secundarios que el inhibidor puede causar sobre nuestro organismo.
Los plazos pueden acortarse muy significativamente si el inhibidor es un fármaco ya aprobado para el tratamiento de otras enfermedades. Esta es la razón que explica el porqué se están realizando pruebas masivas in vitro e in vivo de diferentes fármacos para inhibir alguna de las proteínas del SARS-CoV-2, incluyendo la proteasa. Así, el grupo de Shanghai ha publicado recientemente una estructura de la proteasa principal del SARS-CoV-2 inhibida por carmofur, un antineoplásico utilizado para combatir cánceres colorrectales y que podría ser un punto de partida prometedor para el desarrollo de nuevos antivirales. Cuando podremos disponer de ellos solo la ciencia (y su financiación) lo dirá.
Representación del ataque nucleofílico del Sγ (en amarillo) de la cisteína-145 de la proteasa 3CL sobre el enlace peptídico de la poliproteína sustrato (la dirección del ataque se ha destacado como línea de puntos). Previamente, la histidina-41 ha transferido un átomo de hidrógeno (representado en blanco y con una línea de puntos) al átomo de nitrógeno del enlace peptídico que va a romperse. El vídeo es cortesía de Carlos A. Ramos-Guzmán, investigador de la Universitat de València.